Structure refinement of the OpcA adhesin using molecular dynamics
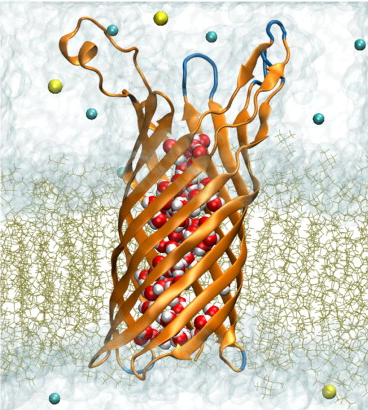
OpcA from Neisseria meningitidis, the causative agent of meningococcal meningitis and septicemia, is an integral outer membrane protein that facilitates meningococcal adhesion through binding the proteoglycan receptors of susceptible cells. Two structures of OpcA have been determined by x-ray diffraction to 2 A resolution, revealing dramatically different conformations in the extracellular loops--the protein domain implicated in proteoglycan binding. In the first structure, a positively charged crevice formed by loops 1 and 2 was identified as the site for binding proteoglycans, whereas in the second structure the crevice was not evident as loops 1 and 2 adopted different conformations. To reconcile these results, molecular-dynamics simulations were carried out on both structures embedded in a solvated lipid bilayer membrane. Free of crystal contacts and crystallization agents, the loops were observed to undergo large structural transformations, suggesting that the conformation of the loops in either x-ray structure is affected by crystallization. Subsequent simulations of both structures in their crystal lattices confirmed this conclusion. Based on our molecular-dynamics trajectories, we propose a model for OpcA that combines stable structural features of the available x-ray structures. In this model, all five extracellular loops of OpcA have stable secondary structures. The loops form a funnel that leads to the base of the beta-barrel and that includes Tyr-169 on its exposed surface, which has been implicated in proteoglycan binding.